
This AI model judges molecular stability on its own, researchers say

A team of Korean researchers led by Professor Woo Youn Kim in the Department of Chemistry in the Korea Advanced Institute of Science and Technology (KAIST) have developed an artificial intelligence model that, they say, understands the physical laws governing molecular stability to predict structures, having published an article on their research in Nature Computational Science (doi.org/10.1038/s43588-025-00919-1).
While existing AI models simply mimic the shape of molecules, the most significant feature of Riemannian DenoisingModel (R-DM) is that it directly considers the ‘energy’ of the molecule, refining the structure by considering the forces acting within the molecule. The molecular structure has been represented by the researchers as a map — where higher energy is depicted as hills and lower energy as valleys, designing the AI to move toward and find the valleys with the lowest energy.
By navigating this energy landscape, avoiding unstable structures to find the most stable state, R-DM completes the molecule. The mathematical theory of Riemannian geometry is applied here, resulting in the AI learning the fundamental law of chemistry: ‘matter prefers the state with the lowest energy’.
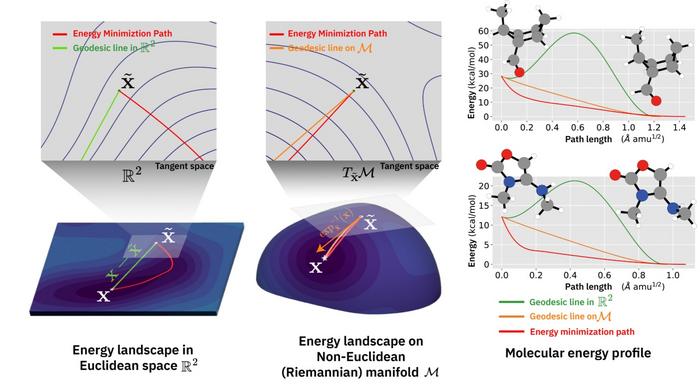
The team says that experimental results have shown R-DM achieved up to 20 times higher accuracy than existing AI models, reducing prediction errors to a level nearly indistinguishable from precise quantum mechanical calculations. This represents, the researchers claim, the world’s highest level of performance among AI-based molecular structure prediction technologies.
“This is the first case where artificial intelligence has understood the basic principles of chemistry and judged molecular stability on its own,” Kim said. “It is a technology that can fundamentally change the way new materials are developed.” High-performance catalyst design, next-generation battery materials and new drug development are among the areas in which this technology can be utilised, the researchers say.

The model is expected to serve as an ‘AI simulator’ that will dramatically speed up research and development by significantly shortening the molecular design process. The team also sees significant potential in environmental and safety fields, as it can quickly predict chemical reaction paths in situations where experiments are difficult, such as chemical accidents or the spread of hazardous substances.
Robotic laboratory to fast-track drug invention, trials and commercialisation
The lab will house an automated system for high-throughput X-ray crystallography, often referred...
Bio-inspired robotic bird has stable flight in its sights
The nankeen kestrel is among the most stable fliers in the avian world. Using motion capture...
Protecting particles that carry mRNA in dry vaccine patches
Combining expertise from RMIT University, MIT and Harvard Medical School, scientists set out to...







